Navigating Life in Three Dimensions: An Introduction to Spatial Biology
Spatial biology is revolutionizing biomedical research by emphasizing the 3-dimensional relationships and arrangements of biological molecules within tissues and [...]

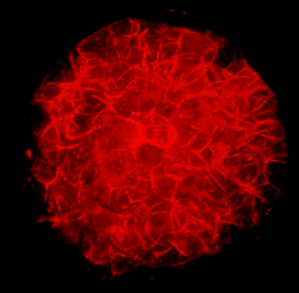
Spatial biology is revolutionizing biomedical research by emphasizing the 3-dimensional relationships and arrangements of biological molecules within tissues and [...]

Ensuring data's reliability, integrity, and accuracy is paramount in scientific research and development. This is where Good Laboratory Practice [...]
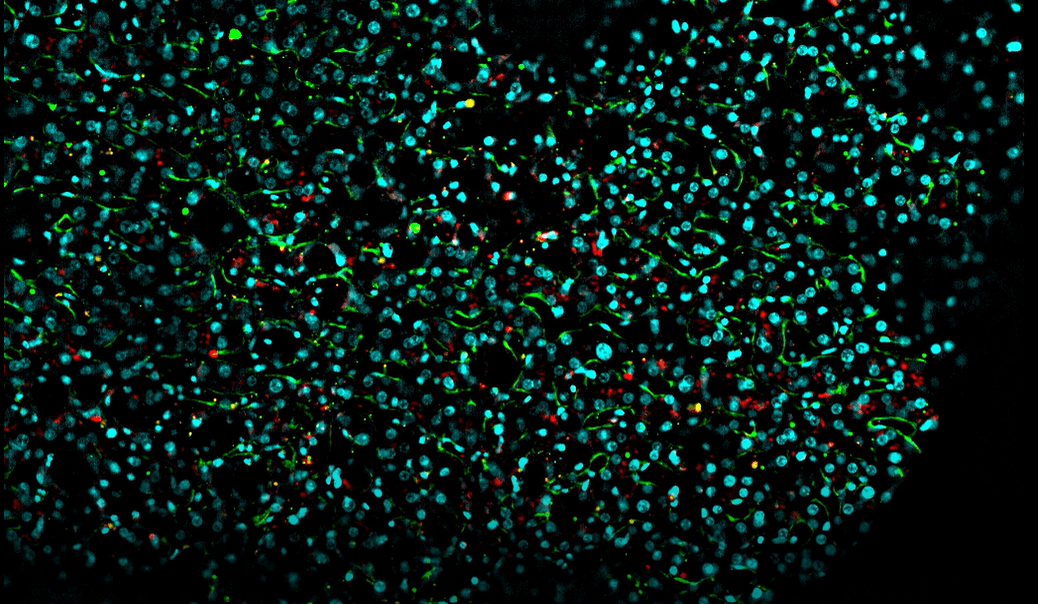
In biological research, whole mount imaging allows for the comprehensive visualization of specimens, revolutionizing our grasp of complex biological [...]
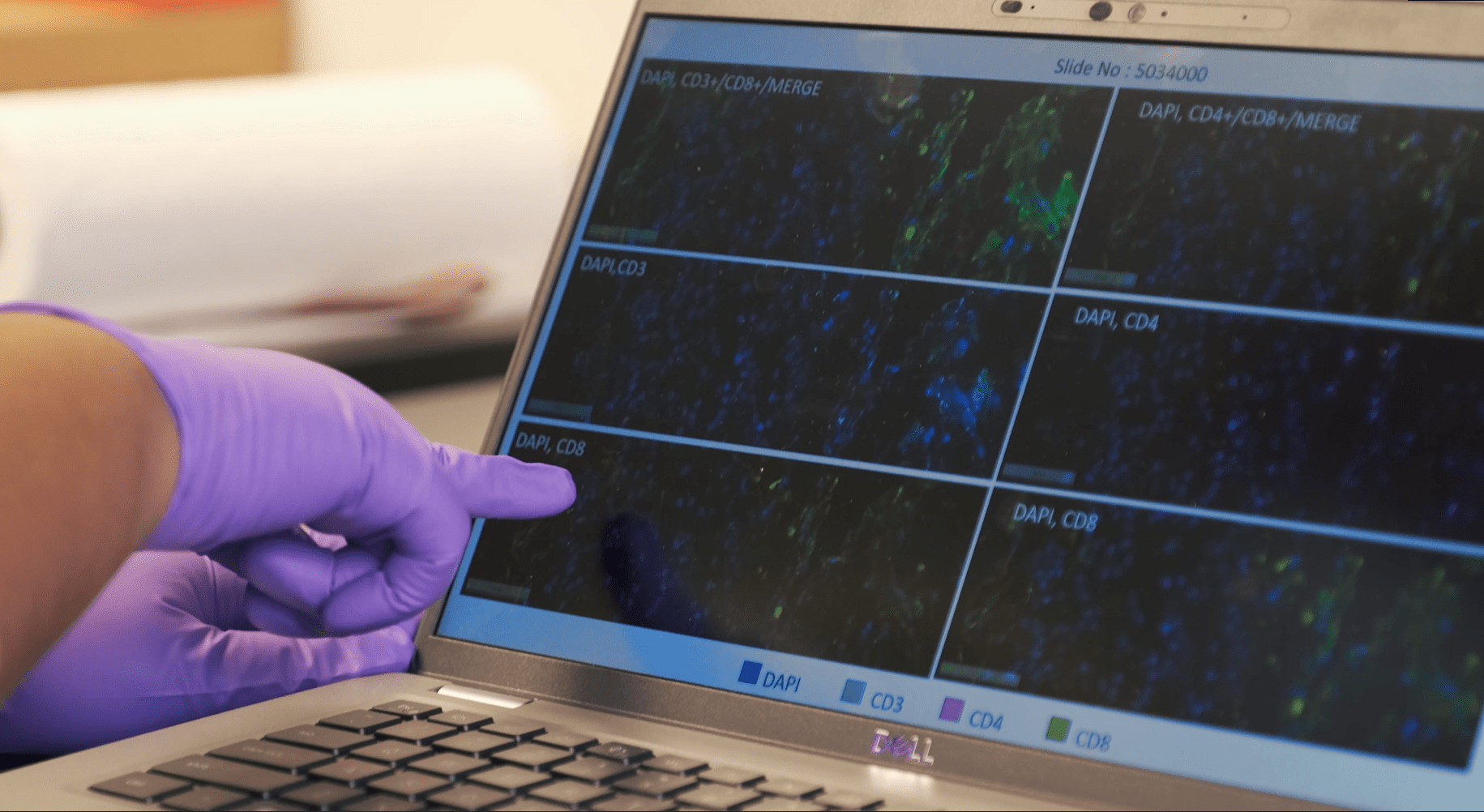
Artificial intelligence (AI) is a broad field defined by the development of machines to perform tasks that would normally [...]
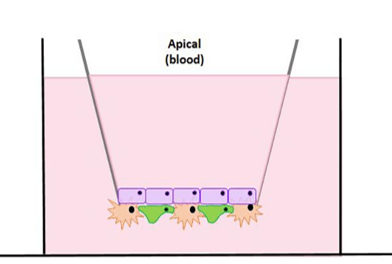
The brain is the central control system of our bodies, which is why it is so heavily protected by [...]
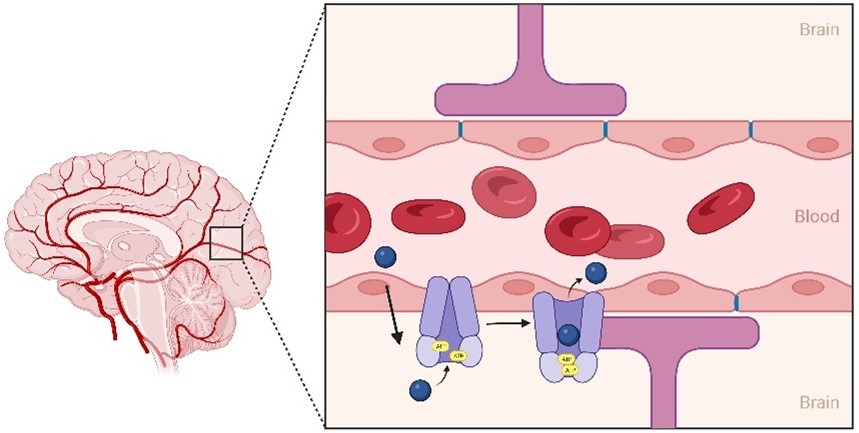
At Visikol, we have pioneered and optimized an in vitro model of the blood-brain barrier (BBB) utilizing a triculture [...]
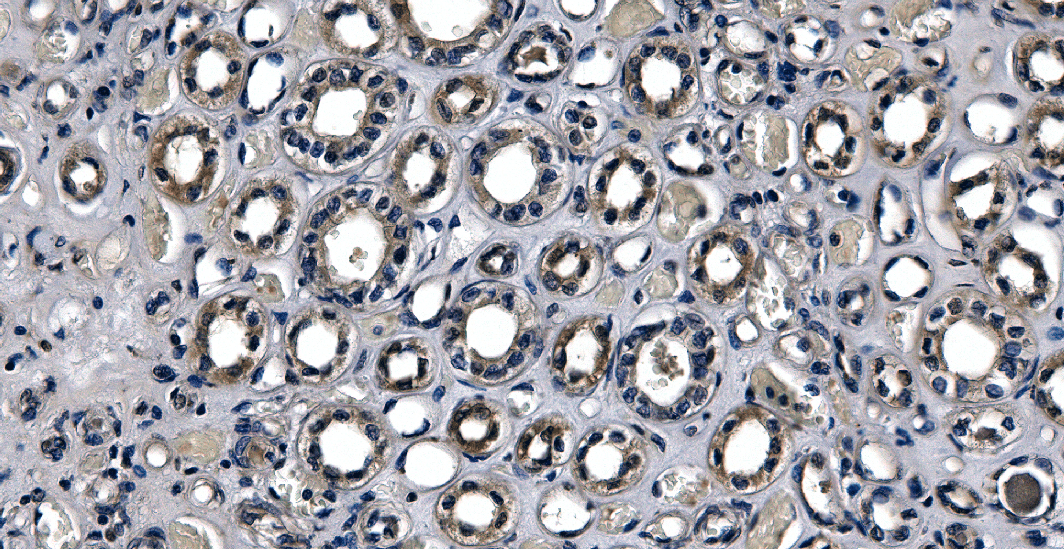
Immunofluorescence, a powerful technique in the realm of histological procedures, allows researchers to visualize and study cellular structures with [...]
While the pathological hallmarks of Alzheimer's disease (AD), a neurodegenerative disorder, have been established, there is ongoing research into [...]
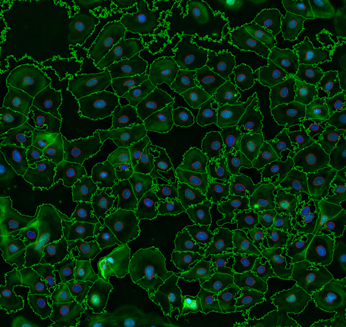
In the quest to unravel the intricacies of cellular biology, scientists have embarked on a journey into the microscopic [...]
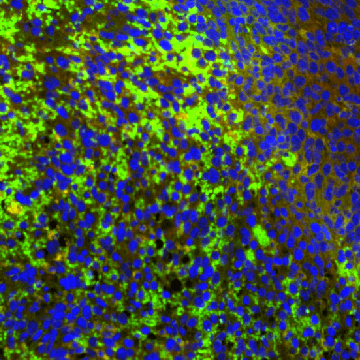
Proteins exhibit astonishing diversity, enabling them to carry out myriad functions within living organisms. Within the human body are [...]