
With the development of tissue clearing technology, the wide availability of fluorescent probes and fluorescent-tagged antibodies, and advances in confocal imaging and light sheet imaging techniques, 3D microscopy for tissue specimens has become a powerful tool for interrogating biology. In this post, we will discuss how to conduct image processing on the resultant image stacks obtained from using these advanced microscopy techniques, using the freely available, open-sourced, and hugely extensible ImageJ imaging platform (available for download here). We recommend using the Fiji package (Fiji Is Just ImageJ), a packaged version of ImageJ pre-loaded with many useful plugins and tools (available for download here).
Confocal Microscopy
Most major universities and institutions have confocal microscopes available for use by researchers. Confocal microscopy is a specialized form of fluorescent microscopy which makes use of a confocal lens apparatus in combination with a pinhole aperture (typically 40-70 μm) in order to screen out light from outside the focal plane (see Figure 1). The result is the removal of out-of-focus light, providing a crisp image with the maximal resolution possible for the objective lens being used. The narrowness of the optical depth combined with the ability to generate successive focal planes by incremental advancement of the focus position between subsequent images, an image stack (also known as a z-stack) can be obtained. Simply put, a z-stack is series of focal planes (along the z axis) at a known interval through a specimen that is
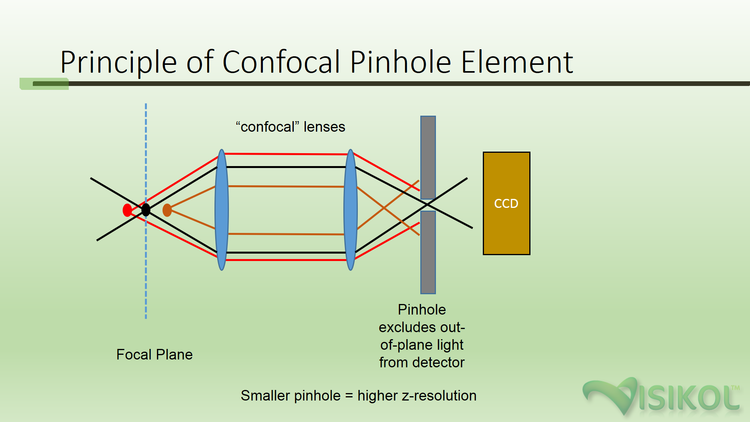
Figure 1. Principle of Confocal Pinhole Element – employing the use of two consecutive lenses, the “confocal lenses,” in the correct geometry, in conjunction with a narrow pinhole, out-of-plane light can be excluded from the detector (CCD).
in a fixed position, providing a three-dimensional sampling of the tissue being examined. This is commonly known as “optical sectioning” wherein thick tissue sections are employed and utilizing the optical ability of the confocal microscope as an optical “microtome,” structural details of the tissue can be revealed in three-dimensions.
The selection of microscope objective for imaging can have serious impacts on quality of images obtained. For most applications, air objectives work effectively; the highest numerical aperture (NA) available at the magnification required should be selected, as higher NA results in more light entering the objective, which yields higher contrast and signal. For deep tissue imaging (> 2mm), the use of glycerol or water immersion objectives is preferred.
Tissue Clearing
To increase the achievable depth at which confocal microscopes can be utilized to obtain images within tissues, clearing techniques (e.g. Visikol HISTO, CLARITY, 3DISCO) can by employed to render tissue specimens optically transparent. In conjunction with fluorescent labeling techniques, tissue clearing dramatically increases the depths at which confocal microscopy can image into tissue, allowing for large scale reconstructions of tissue histology and morphology on large and thick pieces of tissue (> 1 mm).
Loading and Processing Image Stacks in ImageJ (or Fiji)
Three-dimensional confocal imaging results in a collection of images as planes incrementing along the Z axis, known as an “image stack” (also known as a “z-stack”). Depending on the type of instrument and settings used during acquisition, the image may be a sequence of individual images (typically as .TIF files) where each image is a single z-plane in a single color or as a single file containing all images in the stack (e.g. TIF hyperstack, proprietary formats such as Leica .lif file or Zeiss .czi image file).
ImageJ can load TIF hyperstacks directly by using File > Open menu option or dragging and dropping the TIF hyperstack onto the ImageJ window. TIF sequences can be loaded in ImageJ by using the File > Import > Image Sequence menu option. If the image stack has multiple color channels, the loaded stack should be converted to a hyperstack by using the Image > Hyperstack > Stack to Hyperstack menu option in ImageJ. Using this command opens the dialog shown in Figure 2. Select the order of the loaded stack from the dropdown box. Image stacks are loaded in alphabetical order by filename, and so the order corresponding to channel/z-depth/time must be selected. Typically stacks are saved in the XYCZT order, wherein the sequence of XY images is ordered by sequentially by color channel, then z-depth (and for time-series, time as the last dimension), or in XYZCT order, wherein the sequence of XY images is ordered by z-depth, then color channel

(and for time series, time as the last dimension). Select the correct order, and then enter in the number of color channels (c) and steps in the z-sequence (z).
Calibrating Image Stacks
After image stacks are loaded (and converted to hyperstacks if needed), calibration information should be input for the z-stack. This is accomplished by using the Image > Properties menu command. In the dialog, the x, y, and z resolution should be entered; the x and y resolution are defined by the image acquisition parameters, and the z-step (e.g. z resolution) is typically entered into the microscope when acquiring a stack. This step will ensure that distortions or stretching does not occur when processing the image in 3D, and furthermore will allow for direct measurements of the image to be taken.
Proprietary Image Formats
Proprietary image formats (such as the Leica .lif files, or the Zeiss .czi files) can be imported into ImageJ by using the File > Import > Bioformats command (Fiji has Bioformats installed by default). Bioformats is a powerful plugin released by the University of Dundee and Open Microscopy Environment, and enables loading of dozens of proprietary image formats directly into ImageJ. The first dialog screen (Figure 3) allows you to modify various settings about how Bioformats will load the images from the proprietary format, for example, you can tell Bioformats to split the channels into individual stacks (Split channels checkbox), or to automatically stitch tiles of a multi-field image together (Stitch tiles checkbox). For multichannel images, it is convenient to load the stack as a hyperstack, which will automatically separate channel frames and provide a slider bar to select the channel of interest. This can be accomplished by selecting the “Hyperstack” option from the “View stack with” dropdown box at the top left of the dialog. Select all required settings and then click OK.

Figure 3. Bioformats import options dialog pops up when loading proprietary image formats in ImageJ
If the proprietary image format stores multiple image sets in the same file, the Bioformats plugin will show you an additional dialog allowing you to select which image acquisitions you would like to load. Please note that Bioformats will load each checked image set into memory, and if image stacks are each very large, it may consume a lot of memory on your system, which can seriously effect the performance of your computer. Select the image acquisition you would like to load, and click OK.
Measurement of Object Volume from Image Stacks
ImageJ has numerous tools for measuring objects from images, however most of these tools are built to process a single image and return a single result. In order to measure volumes or to repeat measurements across the entire stack, some simple macro programming is required. ImageJ is also very powerful and there are often many ways methods that can be used to accomplish the same goals. In this example, we will demonstrate how to measure the volume of the cytoplasm of a cell, working with the confocal stack sample image that can be opened in ImageJ by going to File > Open Samples > Confocal Series menu option.
Step 1. Split the channels
- The color channels need to be split apart so that we can measure only the cytoplasm channel. This can be accomplished by using the Image > Color > Split Channels menu option. The result is the individual channels in separate windows. Close the red channel since it won’t be needed. Select the green cytoplasm channel.
Step 2. Threshold the Image
- Thresholding is a procedure used in image processing to select pixels of interest based on the intensity of the pixel values. Essentially, thresholding provides for a binary cut-off, wherein pixels above or below the defined threshold are included in analysis, and the rest are ignored. This is very useful for excluding background pixels, selecting bright spots or regions, and selecting objects of interest based on their pixel intensities.
- Select the green channel window, and then select Image > Adjust > Threshold (or hit Ctrl+Shift+T) to bring up the Threshold dialog. The pixels that will be included based on the threshold parameters are shown in red on the image.
- You can assign an automatic threshold method, which determines the threshold value based on a variety of functions that can be

Figure 4. Thresholding dialog box in ImageJ. An automatic threshold method may be selected from the dropdown box (blue outline) which will calculate a threshold value to use for the image. Alternatively, manual thresholding can be done by entering minimum and maximum pixel values (green outline).
- selected from the Threshold method dropdown box (outlined in blue in Figure 4), or you may manually threshold by entering the min and max values you would like to include.
- Select the Otsu method from the automatic threshold dropdown box. The Otsu threshold method is very useful for separating foreground objects from background in microscopy. The Otsu method works by grouping all pixels into two classes, bright and dark, and then determining the average pixel value within each class, then assigning the threshold value as the midpoint between the averages of the bright and dark pixels. This is an effective way to threshold foreground objects from the background in most fluorescent images.
Step 3. Set Measurements
- The next step is to configure the measurements we want to use to measure the thresholded object.
- Select Analyze > Set Measurements menu option from ImageJ. The Set Measurements dialog will appear. Select Area, Mean gray value, Stack position, and Limit to Threshold checkboxes.
Step 4. Simple macro to measure each image of the stack and calculate the volume
- ImageJ was not built to include methods for taking measurements of entire image stacks, or for calculating volumes, and so we’ll need to prepare a very simple macro to accomplish the required measurements.
- The macro code is shown below:

- The macro will loop through each image in the stack, and take the measurement of each slice individually.
- Finally, the macro will loop through and sum the area measurement measurements, multiply this sum by the depth of each slice, and finally display the result.
- There are detailed comments as to what each portion of code does below within comments in the macro code (comments start with // )
- Create a new macro by selecting Plugin > New > Macro menu item in ImageJ, and paste the code below into the macro window.
- Run the macro by clicking the “Run” button at the bottom of the macro window
// ImageJ Macro Code // Measure Volume of Thresholded Pixels in an Image Stack // // macro "Measure Stack" { run("Clear Results"); // First, clear the results table // loop through each slice in the stack. Start at n=1 (the first slice), // keep going while n <= nSlices (nSlices is the total number of slices in the stack) // and increment n by one after each loop (n++) for (n=1; n<=nSlices; n++) { setSlice(n); // set the stack's current slice to n run("Measure"); // Run the "Measure" function in ImageJ } // Create a variable that we will use to store the area measured in each slice totalArea = 0; // Loop through each result from 0 (the first result on the table) to nResult (the total number of results on the table) for (n=0; n < nResults; n++) { totalArea += getResult("Area",n); // Add the area of the current result to the total } // Get the calibration information from ImageJ and store into width, height, depth, and unit variables. // We will only be using depth and unit getVoxelSize(width, height, depth, unit); // Calculate the volume by multiplying the sum of area of each slice by the depth volume = totalArea*depth; // Print the result of the volume calculation to the log print(volume + " " + unit + "^3"); }
Results
If all the code was entered properly, you should see the following windows displayed after the macro is completed (shown in Figure 6). The macro correctly calculated the total volume as 69.6598 µm^3
To run this macro on other images in the future, just save the macro to your hard drive, and it can be run from ImageJ by selecting the Plugins > Run > Macro menu item. Be sure the image you wish to measure was selected prior to running the macro. Images should be single-channel, otherwise the macro will calculate the total volume of thresholded objects across all channels. Images should also be calibrated, so that the proper units are used for calculations.
As can be easily seen, ImageJ is a very powerful platform for image analysis, and with its full scripting language, it is easy to command ImageJ functions in many ways to accomplish a variety of image processing tasks.

Download Tissue Clearing Ebook
If you are interested in learning more about tissue clearing, download our free ebook below which details how best to implement tissue clearing into your research workflow and which tissue clearing approach is best for your specific research question.
